Abstract
Background: The anti-epileptic NKCC1 inhibitor Bumetanide (BUM) and the microtubule acting anthelminthic agent Mebendazole (MEB), have anti-cancer properties. Tumor & its environment generate neuronal hyperactivity that aggravates the clinical outcome suggesting that their combination might block hyperactivity (BUM) and augment cell death (MEB). Methods: We tested the effects of the combo on i) NKCC1 activity in human cell lines, ii) electrical activity recorded from mouse hippocampal neurons & tumors freshly resected from patients, iii) glioblastoma-brain co-cultures, iv) cell death in human tumoroids. Results: BUM efficiently inhibited NKCC1 & unexpectedly, MEB applications also via a likely indirect action. In rodent hippocampal neurons, BUM blocked GABAergic Giant Depolarizing Potentials and seizures and co-applications of MEB produced a fourfold increase of BUM's efficacy. In freshly removed brain tumors, E GABA reversal recorded with single GABA channels was highly depolarized (close to -25 mV) in keeping with NKCC1 over activity. BUM fully blocked ongoing epileptic activity. In GBM-Brain cultures, the combo produced stronger effects then independent applications of MEB or BUM. In tumoroids, the combo also efficiently produced strong cell death & morphological changes in some tumors. Conclusion: The combination of BUM & MEB acts complementarily on brain tumors, the former blocking seizures, and the latter producing cell death. Their combination increases their hyperactivity inhibitory actions and cell death. The combo might therefore be used to treat brain tumors combining 2 different mechanisms and targets.
|
Published in
|
Cancer Research Journal (Volume 13, Issue 3)
|
|
DOI
|
10.11648/j.crj.20251303.12
|
|
Page(s)
|
98-122 |
|
Creative Commons
|

This is an Open Access article, distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is properly cited.
|
|
Copyright
|
Copyright © The Author(s), 2025. Published by Science Publishing Group
|
Keywords
Brain Tumors, Anthelminthic, NKCC1, Bumetanide, Treatment
1. Introduction
In spite of extensive efforts and investments, Glioblastoma (GBM), grade II/III meningiomas and other aggressive brain tumors (BT) remains orphan of novel therapies to ameliorate the life of patients and increase their survival
| [1] | Yabo, Y. A., Niclou, S. P. & Golebiewska, A. Cancer cell heterogeneity and plasticity: A paradigm shift in glioblastoma. Neuro. Oncol. 24, 669-682 (2022). |
| [2] | Stupp, R. et al. Maintenance therapy with tumor-Treating fields plus temozolomide vs temozolomide alone for glioblastoma a randomized clinical trial. JAMA - J. Am. Med. Assoc. 314, 2535-2543 (2015). |
| [3] | White, J., White, M. P. J., Wickremesekera, A., Peng, L. & Gray, C. The tumour microenvironment, treatment resistance and recurrence in glioblastoma. J. Transl. Med. 22, 1-14 (2024). |
| [4] | A Roncevic, N Koruga, A S Koriga, R. R. Why do Glioblastoma treatments fail? Futur. Pharmacol. 5, 1-16 (2025). |
[1-4]
. Reference therapy is the association of Temozolomide (TMZ) and radiotherapy, but prognosis remains low with a median survival of 15 months. Combined with Temozolomide, applications of low intensity alternating electric fields on transducer arrays placed on the skin above the region containing the BT increased survival by 4 to 6 months
| [5] | Stupp R et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma A Randomized Clinical Trial. 318, 2306-2316 (2017). |
[5]
. This technique is however limited by local side effects, short additional survival and high cost.
Inhibitors of the co-transporter NKCC1 exert anti-cancer activity. NKCC1 is highly expressed on the leading edge of cell migration
| [6] | Garzon-Muvdi, T. et al. Regulation of brain tumor dispersal by NKCC1 through a novel role in focal adhesion regulation. PLoS Biol. 10, (2012). |
[6]
. Blocking NKCC1 with BUM or NKCC1 KO attenuates GBM expansion
| [7] | Algharabil, J. et al. Inhibition of Na-K + -2Cl - cotransporter isoform 1 accelerates temozolomidemediated apoptosis in glioblastoma cancer cells. Cell. Physiol. Biochem. (2012) https://doi.org/10.1159/000339047 |
[7]
. Bumetanide slows cancer cell migration
| [8] | Kourghi, M., Pei, J. V., De Ieso, M. L., Flynn, G. & Yool, A. J. Bumetanide derivatives AqB007 and AqB011 selectively block the aquaporin-1 ion channel conductance and slow cancer cell migration. Mol. Pharmacol. 89, 133-140 (2016). |
[8]
and accelerates temozolomide induced apoptosis of GBM cancer cells
| [7] | Algharabil, J. et al. Inhibition of Na-K + -2Cl - cotransporter isoform 1 accelerates temozolomidemediated apoptosis in glioblastoma cancer cells. Cell. Physiol. Biochem. (2012) https://doi.org/10.1159/000339047 |
[7]
. NKCC1 activity is involved in the epithelial to mesenchymal transition
| [9] | Sun, H., Long, S., Wu, B., Liu, J. & Li, G. NKCC1 involvement in the epithelial-to-mesenchymal transition is a prognostic biomarker in gliomas. PeerJ 8, 1-14 (2020). |
[9]
and the spread of cancer cells is abolished in a nude mouse intracranial modelNKCC1 KO
The efficacy of NKCC1 inhibitors to restore GABAergic inhibition in many brain disorders has extensively been shown including in clinical trials
| [11] | Ben-Ari, Y. NKCC1 Chloride Importer Antagonists Attenuate Many Neurological and Psychiatric Disorders. Trends Neurosci. 40, 536-554 (2017). |
| [12] | Savardi, A., Borgogno, M., De Vivo, M. & Cancedda, L. Pharmacological tools to target NKCC1 in brain disorders. Trends Pharmacol. Sci. 42, 1009-1034 (2021). |
| [13] | Lemonnier, E. et al. Effects of bumetanide on neurobehavioral function in children and adolescents with autism spectrum disorders. Transl. Psychiatry 7, 1-9 (2017). |
[11-13]
.
Other observations suggest that the anthelminthics Mebendazole (MEB) and Albendazole (ALB) have powerful antiproliferative, proapoptotic actions with Ic50 below micromolar range in vitro and in vivo
| [14] | Florio, R. et al. Screening of benzimidazole-based anthelmintics and their enantiomers as repurposed drug candidates in cancer therapy. Pharmaceuticals 14, 1-16 (2021). |
[14]
They have relatively good brain penetration
| [15] | Bai, R. Y., Staedtke, V., Aprhys, C. M., Gallia, G. L. & Riggins, G. J. Antiparasitic mebendazole shows survival benefit in 2 preclinical models of glioblastoma multiforme. Neuro. Oncol. 13, 974-982 (2011). |
[15]
, inhibit tubulin polymerization, pro-survival pathways, matrix -metalloproteinases, angiogenesis, drug resistant protein transporters and attenuate several other types of cancers and tumors
| [16] | Simbulan-rosenthal, C. M. et al. The repurposed anthelmintic mebendazole in combination with trametinib suppresses refractory NRAS Q61K melanoma. 8, 12576-12595 (2017). |
| [17] | Williamson, T. et al. Mebendazole disrupts stromal desmoplasia and tumorigenesis in two models of pancreatic cancer. Oncotarget 12, 1326-1338 (2021). |
[16, 17]
. Promising results have recently been obtained in GBM phase 1-2 clinical trials using MEB
| [18] | Gallia, G. L. et al. Neuro-Oncology Advances diagnosed high-grade gliomas : results of a phase 1. neuro-oncology Adv. 3, 1-8 (2021). |
[18]
.
Recent observations suggest that the environment of the tumors plays an important role in its aggravation, with functional GABAergic and glutamatergic synapses established associated with network hyperactivity that aggravate its severity
| [19] | Losada-Pérez, M., García-Moreno, M. H., García-Ricote, I. & Casas-Tintó, S. Synaptic components are required for glioblastoma progression in Drosophila. PLoS Genet. 18, 1-27 (2022). |
| [20] | Venkatesh, H. S. et al. Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature (2017) https://doi.org/10.1038/nature24014 |
[19, 20]
. Indeed, blocking this neuronal activity attenuates metastasis
| [19] | Losada-Pérez, M., García-Moreno, M. H., García-Ricote, I. & Casas-Tintó, S. Synaptic components are required for glioblastoma progression in Drosophila. PLoS Genet. 18, 1-27 (2022). |
| [21] | Wang, X., Liang, J. & Sun, H. The Network of Tumor Microtubes: An Improperly Reactivated Neural Cell Network With Stemness Feature for Resistance and Recurrence in Gliomas. Front. Oncol. 12, 1-11 (2022). |
[19, 21]
. In tumors, GABA exerts depolarizing actions due to high (Cl
-)
i levels
Collectively, these observations stress the importance to target in a therapeutic perspective the tumor AND its environment.
Here, we tested the hypothesis that Meb and BUM might have complementary actions; the former leading to apoptosis and the latter blocking environmental hyperactivity. We report that BUM & MEB have complementary actions on brain GBM cocultures, Tumoroids of GBM and meningiomas. A synergistic action is conspicuous in rodent brain slices, where co-applications of MEB with BUM reduced significantly BUM dosage needed to restore GABAergic inhibition. Collectively, results suggest that the combo might constitute a novel approach to treat GBM and aggressive brain tumors.
2. Material & Methods: Also See Complementary M&M
1. Determining NKCC1 inhibition in cell lines
NKCC1 function was assessed in HEK293 cells as previously described
| [23] | Koumangoye, R., Penny, P. & Delpire, E. Loss of NKCC1 function increases epithelial tight junction permeability by upregulating claudin-2 expression. Am. J. Physiol. - Cell Physiol. 323, C1251-C1263 (2022). |
[23]
. HEK293 cells were first exposed to an isosmotic saline, followed by an uptake with an identical saline containing 200 mM ouabain, 0.25 mCi/mL
83Rb, and drugs. Mebendazole was tested in concentrations ranging from 100 pM to 31.6 mM in the presence or absence of 20 mM bumetanide. K
+ influx was calculated from
83Rb tracer uptake and expressed in pM K
+ x mg protein
-1 x min
-1.
2. Electrophysiology
1) Rodent Hippocampal Slice preparation
P4-P15 mice hippocampal slices were prepared as previously described
| [24] | Lozovaya, N. et al. Early alterations in a mouse model of Rett syndrome: the GABA developmental shift is abolished at birth. Sci. Rep. 9, 1-16 (2019). |
[24]
& incubated in artificial cerebrospinal fluid.
Dosages of BUM and MEB
In our electrophysiological investigations on rodent and human in vitro material, we relied on the dosages used in the extensive in vitro studies made with BUM of MEB usually 10uM BUM on slices known to restore GABAergic inhibition in a wide range of studies
| [25] | Ben-Ari, Y., Gaiarsa, J.-L., Tyzio, R. & Khazipov, R. GABA: A pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol. Rev. 87, 1215-1268 (2007). |
| [11] | Ben-Ari, Y. NKCC1 Chloride Importer Antagonists Attenuate Many Neurological and Psychiatric Disorders. Trends Neurosci. 40, 536-554 (2017). |
[25, 11]
. For MEB, as very few studies have been done on slices, we used 10 or 20uM found not to have effects on intrinsic currents and as its actions are meant to be on microtubules, we incubated the slices in its presence for 30 minutes to 1-2 hours.
2) Human GBM acute slice preparation
After surgical resection, the brain tissue was placed within 30 s in ice-cold oxygenated protecting solution that contained in (mM): 110 choline chloride, 26 NaHCO3, 10 D-glucose, 11.6 sodium ascorbate, 7 MgCl2, 3.1 sodium pyruvate, 2.5 KCl, 1.25 NaH2PO4 and 0.5 CaCl2, 300 mOsm and brought to the neurophysiology laboratory within <10 min. Brain slices (300-400 μM) were prepared and transferred to the holding chamber in which they were stored at room temperature (20-22 °C) in ACSF containing in mM): 125 NaCl, 3.5 KCl, 0.5 CaCl2, 3 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, and 10 glucose, 300 mOsm, equilibrated at pH 7.3 with 95% O2 and 5% CO2 at room temperature (RT) (22-25 °C) for at least 2 h to allow recovery. For recordings, we used the same solution but with 2 mM CaCl2 and 1 mM MgCl2.
3) Patch-clamp recordings in rodent and human brain slices
Slices were perfused with oxygenated recording ACSF at 3 ml/min-1 at RT. Neurons were visualized using infrared differential interference contrast microscopy. Patch pipettes (7-9 MΩ resistance) where filled with intracellular solution (in mM): 130 K-gluconate, 10 Na-gluconate, 7 NaCl, 4 MgATP, 4 phosphocreatine, 10 HEPES, and 0.3 GTP (pH 7.3 with KOH, 280 mOsm). Biocytin (final concentration 0.3-0.5%) was added to the pipette solution to label neurons in human GBM slices. Cells with leakage current more than 40 pA were discarded.
To determine cell excitability, we recorded voltage responses (current-clamp mode) to 1 s current steps of − ep to + 150 pA (10 pA increment, 3 s interval between each step). Current-voltage (I-V) relationships were established to calculate input resistance of cells. Action potential duration (half-width) was measured at half of the maximal amplitude. Spontaneous excitatory postsynaptic currents (EPSCs) were recorded for 10 min at −t min and spontaneous GABAergic postsynaptic currents (GPSCs) at 0 mV (the reversal potential for glutamatergic currents). Membrane potentials were corrected for a liquid junction potential of −ft mV (calculated using the junction potential calculator module of pClamp 10). Extracellular tetanus stimulation
To induce seizure-like events (SLE), a bipolar Ni-Cr electrode was positioned on the surface of hippocampal slices close to the recorded cell. Trains (20 stimuli, 100 Hz) of current pulses (25-50 μA, 100 µs) were delivered every 120 s through the constant current bipolar stimulus isolator A365 (World Precision Instruments).
4) Single-channel recordings from GBM cells in human brain slices
Patch pipette solution for single GABAA channels recordings contained (in mM): NaCl 120, KCl 5, TEA-Cl 20, 4-aminopyridine 5, CaCl2 0.1, MgCl2 10, glucose 10, Hepes 10 buffered to pH 7.2-7.3 (pipette resistance 7-10 MΩ) & 5 μM GABA. Cell-attached recordings were made under visual control. After gigaseal formation (> 3 GΩ) currents through GABAA channels of 1 pA were immediately visible at the potential + 50 mV. Currents through GABAA channels were recorded from − om mV to + 50 mV with 10 mV increments for 1-2 minutes for each holding potential. We performed analysis of single channel currents and I-V relationships using Clampfit 10.6 (Axon Instruments, Union City, CA).
3. Glioblastoma-Brain culture and assays
Glioblastoma cells were cultured as previously described by
| [26] | Martinez Bedoya, D. et al. PTPRZ1-Targeting RNA CAR T Cells Exert Antigen-Specific and Bystander Antitumor Activity in Glioblastoma. Cancer Immunol. Res. 12, 1718-1735 (2024). |
[26]
. GE835 cells were transduced with UBI-fLUC-PGK-mCherry lentivirus and selected by mcherry expression. Neural organoids (or neurospheres) were differentiated from human iPSCs (Reprocell). IPSCs were transduced with UBI-GFP lentivirus and selected by GFP expression. The protocol used for differentiation of iPSCs in neurospheres was slightly modified from
| [27] | Cosset, É. et al. Human three-dimensional engineered neural tissue reveals cellular and molecular events following cytomegalovirus infection. Biomaterials 53, 296-308 (2015). |
[27]
. Imaging and analysis of Neuro
Gfp GBM
mcherry/fLuc co-culture, Neuro
Gfp glio
mcherry/fLuc culture dissociation, Draq7 staining and FACS analysis, apoptosis, neural differentiation quality control (RNA extraction and qPCR), and spheroid viability assay with 3D CellTiter-Glo are detailed in the supplementary data.
4. Tumoroid Formation and Culture
Tumoroids were generated from patient-derived tissue samples obtained through biopsy under ethically approved protocols with informed patient consent. Tumoroids were cultivated using a tumoroid-on-chip platform to replicate tumor microenvironment and maintain the three-dimensional (3D) architecture necessary for accurate drug screening.
1) Drug Treatments
BUM and MEB were prepared as DMSO stock solutions and diluted in culture medium to reach working concentrations. Live/Dead Viability Assay, imaging and quantification, and Morphological Analysis are explained in supplementary data.
2) Statistical Analysis
Statistical analysis was conducted using GraphPad Prism 10 (GraphPad Software, USA). Data from triplicates were pooled, and statistical significance was assessed using one-way ANOVA, followed by post-hoc multiple comparisons. Comparisons between treatment groups were performed to evaluate potential synergistic effects.
3) Tumor Sample Collection and Processing
Tumor samples were obtained from patient biopsies of glioblastomas, meningiomas, and metastases, collected under ethically approved protocols with informed patient consent.
4) ECM Matrices
4 different ECM platforms were evaluated: VivoInk (CellInk), Agarose Low Melting (Sigma-Aldrich), Laminink+ (CellInk) and Telocol-6 (CellInk).
3. Results
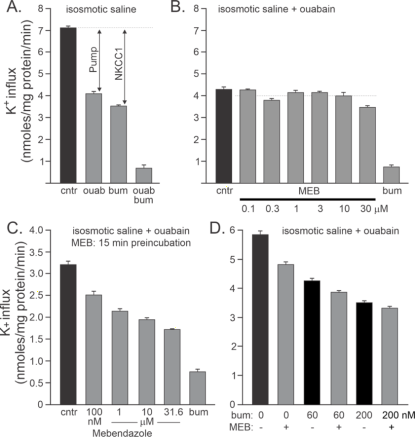
1. Effects of MEB and BUM on K+ fluxes
K
+ influx in HEK293 using
83Rb as a tracer for K
+ movement, most of the K
+ influx is mediated by the Na
+/K
+ pump (ouabain-sensitive component) and the Na-K-2Cl cotransporter (bumetanide-sensitive component) (
Figure 1). We observed that unlike bumetanide, MEB had no direct effect on NKCC1 function (
Figure 1B). However, when used during the pre-incubation and flux periods, MEB decreased NKCC1 function (
Figure 1C). The MEB IC
50 was determined to be 157 (not shown) but yielding only 50% inhibition. When combined with bumetanide, MEB showed effect only at very low doses of bumetanide (
Figure 1D) suggesting different sites of actions & possible indirect interactions of MEB & NKCC1.
2. Complementary Effects of the combo on rodent hippocampal neurons
A. MEB and BUM do not alter intrinsic currents
Applications of MEB (10 or 20 µM), BUM (10 µM), or the MEB analogue Albendazole (ALB 20 µM) produced no change in half-width AP duration, AP threshold, input resistance, spike amplitude in current-clamp whole-cell recordings of CA1 hippocampal pyramidal neurons after 1 h incubation (Suppl
Figure 1). This was not altered by the addition of BUM (10 µM). Similar results were observed with the MEB powerful analogue Albendazole (10 µM) (Suppl
Figure 2).
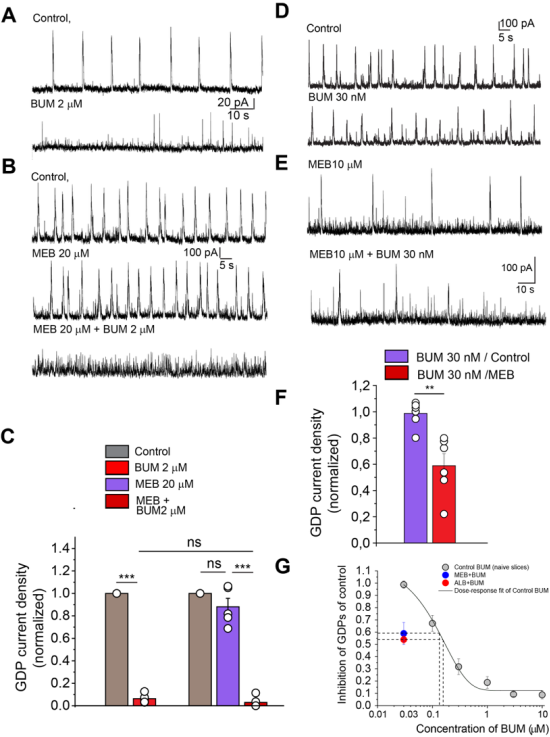
B. Synergistic actions of benzimidazoles and BUM on inhibition of GDP in neonatal hippocampal slices
The immature hippocampus generates spontaneous network-driven synchronous oscillation, Giant Depolarizing Potentials (GDPs)
| [28] | Ben-Ari, Y., Cherubini, E., Corradetti, R. & Gaiarsa, J. L. Giant synaptic potentials in immature rat CA3 hippocampal neurones. J. Physiol. 416, 303-325 (1989). |
[28]
that are mediated largely by depolarizing GABAergic currents that are extremely sensitive to NKCC1 block by bumetanide (Spoljaric et al, 2017). Incubation with Meb (20 µM) did not affect significantly GDP current density in CA3 pyramidal neurons in hippocampal slices (0.88 ± 0.08 of control, n = 5), while bumetanide (2 µM) blocked GDP current density to 0.06 ± 0.02 of control in naïve slices and to 0.03 ± 0.02 of control (n = 5) in MEB-treated slices with no statistically significant differences (
Figure 2A-C). However, in mebendazole treated slices, 30 nM BUM that does not alter GDP in control conditions (0.98±0.03 of control, n=8) produced a significant inhibition (0.71±0.05 of control, n=4) (
Figure 2E, F) (
Figure 3D, F, G). Therefore, mebendazole treatment enhance bumetanide potency.
Similar results were observed with the MEB analogue Albendazole (10 µM) that did not affect significantly GDP activity in CA3 pyramidal neurons (1.21 ± 0.4, n = 4) (Suppl
Figure 2 A, B). BUM (2 µM) fully blocks GDP activity (0.02 ± 0.02 of control (n = 4) in control slices; but BUM (30 nM) which did not alter GDP activity in control conditions (0.98±0.03 of control, n=8) significantly inhibited them (0.54±0.03 of control, n=9) in albendazole-treated slices (Suppl
Figure 2C-E). Thus, benzimidazoles treatment enhance significantly the potency of bumetanide inhibition.
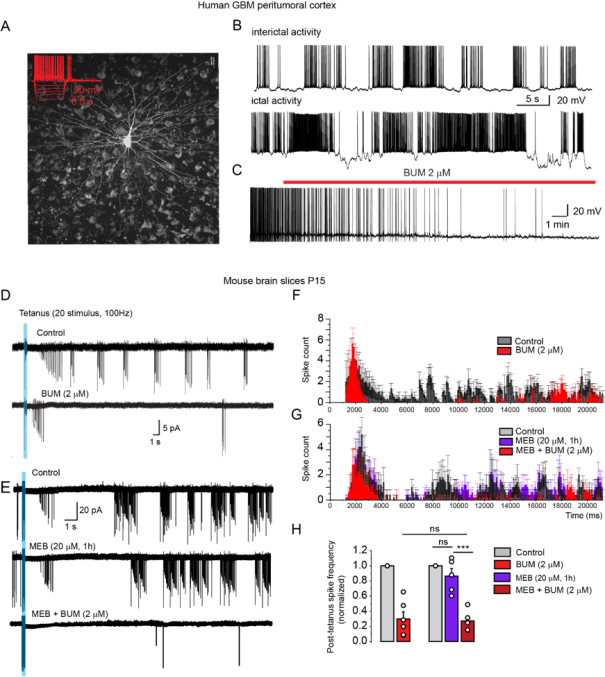
C. Complementary actions of MEB and BUM to suppress epileptiform activity
Spontaneous epileptiform activity was recorded in pyramidal neurons of peritumoral cortex in acute human BT slices (
Figure 3A, B). This activity was fully blocked by bumetanide (2uM) (
Figure 3C).
In p14-p15 mice, seizure-like events (SLE) were triggered in CA1 pyramidal neurons by tetanic stimulation of stratum radiatum (
Figure 3D). Bumetanide (2 µM) significantly decreased post-tetanus spike frequency to 0.30±0.09 of control (n=6, two-tailed, paired t-test, p=7*e-4) in naive slices and to 0.27±0.06 of control (n=5, two-tailed, paired t-test, p=3*e-4) in mebendazole -treated slices (
Figure 3 D-H). MEB alone (20 µM) applied alone for 1 h did not affect SLE (0.86±0.09, n=5) (
Figure 3E, G, H). Similarly, Albendazole application for 1 h did not affect SLE (0.92 ± 0.09, n = 10). But in albendazole-treated slices, Bumetanide (2 µM) significantly decrease post-tetanus spike frequency to 0.32 ± 0.05 of control (n = 10, two-tailed, paired t-test, p = 7*e-4) (Suppl.
Figure 3).
D. Mebendazole and bumetanide combination restored inhibitory driving force of GABA (DFGABA) in GBM cell in peritumoral cortex.
To determine (Cl
-)
i levels and its modulation by combo treatment, we performed non-invasive cell-attached single-channel GABA
AR current recordings in glioma cell of BT infiltrated cortex (Suppl
Figure 4A, B). Pyramidal neuron located close to glioma cell displayed spontaneous glutamatergic hyperactivity (Suppl
Figure 4C). Glioma cells were identified by 5-ALA staining and electrophysiological properties (Suppl
Figure 4D). I/V plot of single-channels GABA
AR currents shows reversal potential close to -25 mV, assuming resting potential of about -40 mV (-38.7 ±2.0, n=7), implying that within the tumors malignant glioma cells have high [Cl
-]
i levels indicative of NKCC1 overactivity (Suppl
Figure 4G, H). 1 hour incubation with mebendazole (10 µM) did not change DFGABA, but BUM (2 µM) decreased DFGABA with a ~20 mV shift of I/V curve to more hyperpolarized potentials (Suppl
Figure 4 H). In addition, glioma cells that generate small action potentials, displayed BUM (2 µM) sensitive ongoing oscillations (Suppl
Figure 5). Collectively, these observations validate the enhanced activity of NKCC1 and the efficacy of BUM to block oscillations, synapse driven hyperactivity including when combined with MEB.
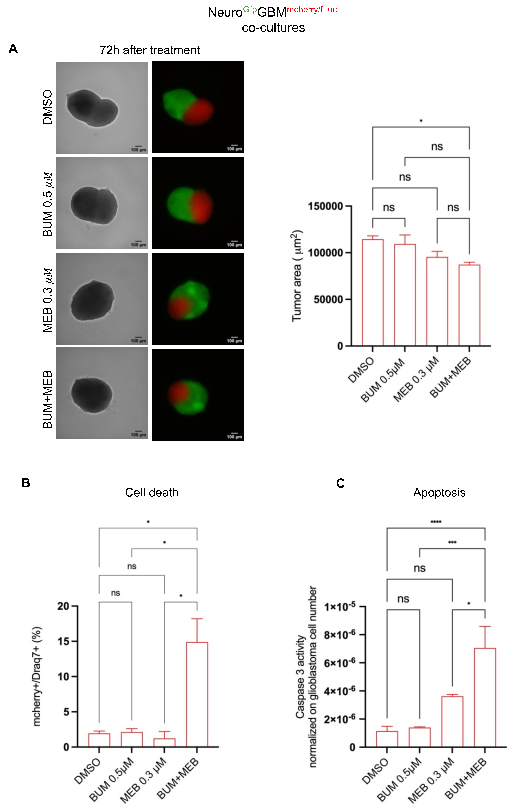
3. Effects of MEB & BUM on NeuroGfp GBMmcherry/fLuc cultures
Co-cultures are made up of patient-derived glioblastomas that grow and invade engineered iPSC-derived neural tissue in a 3D culture. Glioblastoma and neural tissue can be distinguished by the expression of mcherry/fluc (red) reporter and GFP (green), respectively. After five days of co-culture, GBM cells spread from the spheroid invading the neural compartment as well as infiltrating neural cells into the tumor part (Supplementary
Figure 6 A, B).
Before starting the co-culture and treatment, we performed an analysis of neural differentiation markers in neural tissue derived from iPSCs (neuro
Gfp), which indicated that neural markers were expressed after 44 days of differentiation, whereas stemness markers (OCT-4) disappeared, indicating that neurons and glial cells are present (Suppl
Figure 6 C). GBM spheroids and neuro
Gfp were co-cultured for three days, and tumors and neurospheres were measured before treatment, with no differences at time zero (Area T0) (Suppl
Figure 6A, B).
BUM (0.5
M) and MEB (0.3
M) were tested independently or combined. As compared with DMSO, the combination of these drugs significantly reduced tumor area (*p-value=0.05), calculated 72 hours after treatment. Independent applications did not significantly impact tumor area (
Figure 4A) (Supplemental
Figure 7A, B) and neural tissue size was not affected by any condition (supplemental
Figure 7C, D), indicating no neurotoxicity (p-value=not significant) (Suppl
Figure 7C).
Draq7 staining to evaluate cell death, showed that combined treatment significantly increased the percentage of cell death 48 hours after treatment which was significantly higher than DMSO or MEB and BUM alone (
Figure 4B). Draq7 positive cells were also found in neural tissue, in a range of 0.05 to 1.5% that does not significantly differ from DMSO (Suppl
Figure 7D). This may result from the turnover of cells or from dissociation processes during which each culture must pass prior to being subjected to flow cytometry analysis.
Next, the treatment's ability to induce apoptosis was determined using caspase-3 activity normalized to tumor cell number. Co-cultures treated with DMSO, and bumetanide showed low caspase-3 activity. MEB alone increased caspase-3 activity, although not significantly compared to DMSO. Combinations of drugs caused a significant increase in tumor apoptosis compared to DMSO and to independent MEB/BUM applications (
Figure 4 C). Thus, the combo has a synergistic effect on cell death, apoptosis, & tumor growth, without significant neurotoxicity.
4. Effects of MEB & BUM on Tumoroids
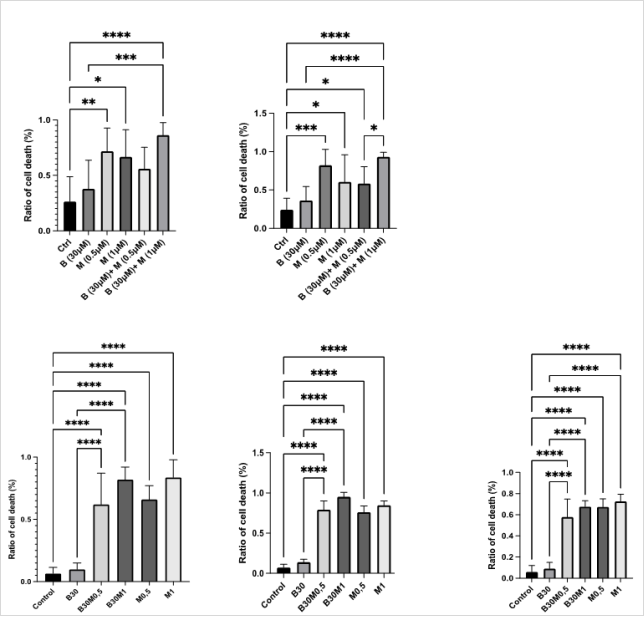
The effects of BUM and /or MEB were tested on a total of 16 tumoroids freshly extracted from patients. In some, both drugs were tested, in other either BUM or MEB were tested because of limited biological material. In
Figure 5, the cytotoxic effects of BUM30 and MEB0.5 and MEB1 were evaluated across tumoroids derived from four meningiomas patients.
Patient1-Determined 24 and 48 hours after surgery (Patient 1- (
figure 5)), the cytotoxic impact of BUM and MEB, individually and in combination showed a progressive increase of cell death. Indeed, the combination treatment BUM30 MEB1 significantly increased cell death compared to control at 24 hours (****p < 0.0001), with a cell death ratio of 0.96 (96%), and this effect was further pronounced at 48 hours, reaching a ratio of 0.99 (99%; ****p < 0.0001).
Patient 2- after 48 hours, the control condition exhibited a significantly lower cell death ratio compared to all treatment conditions (****p < 0.0001) excepted the condition BUM30. MEB0.5 alone produced a cell death ratio of approximately 0.68, while MEB1 increased it to 0.74. However, the combination BUM30 MEB0.5 did not substantially elevate the effect, maintaining a ratio of 0.70. Notably, the BUM30MEB1 combination did not outperform MEB1 alone, showing a slightly lower ratio of 0.73 (****p < 0.0001).
Patient 3- at 48 hours, MEB0.5 achieved a cell death ratio of approximately 0.72 (****p < 0.0001), and MEB1 increased it to 0.80. The combination BUM30 MEB0.5 showed a similar effect with a ratio of 0.81, while BUM30 MEB1 further elevated the response to 0.88 (****p < 0.0001).
Patient 4- the control condition showed significantly lower cell death ratios across all treatment conditions (****p < 0.0001) excepted for the condition BUM30. MEB0.5 produced a ratio of approximately 0.65, MEB1 0.71, BUM30 MEB0.5 0.74, and with BUM30 MEB1 0.84 a consistently increased cytotoxicity producing the highest effect (****p < 0.0001). These findings underscore the variability in tumoroid responses, with Mebendazole as the primary driver of cell death, and Bumetanide displaying a more pronounced effect in combination at higher doses. Taken together, the BUM30 + MEB1 combination emerged as the most consistently effective condition across all four patient-derived meningioma tumoroid models, achieving near-total cell death in each case.
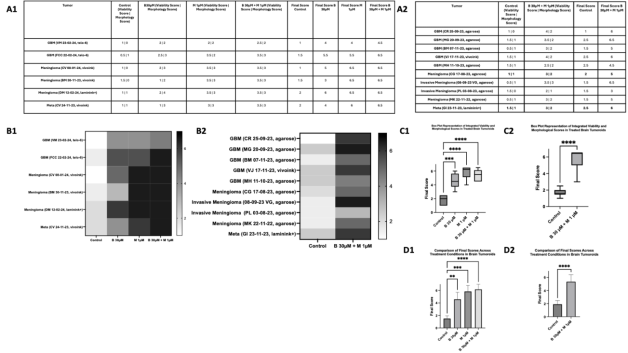
In
Figure 6, the assessment of treatment efficacy is structured across four sections: A1 A2 (quantitative assessment), B1 B2 (heatmap visualization), C1 C2 (box plot distribution), and D1 D2 (bar plot of median final scores). The analysis includes a total of 16 tumoroids, with 10 tumoroids tested under control and BUM30 + MEB1 combination only due to limited tissue availability, and 6 tumoroids assessed under all conditions (control, BUM30, MEB1, BUM30 + MEB1). The final score, representing the combined assessment of viability (cytotoxicity) and morphological changes (cellular integrity), was calculated for each tumoroid. In A1 A2, quantitative assessment reveals that BUM30 + MEB1 consistently achieved the highest final scores across the majority of tumoroids, indicating a pronounced and sustained therapeutic effect. The synergy between BUM30 and MEB1 is particularly evident where the median final score is 6.5, suggesting near-complete cell loss and severe morphological disruption.
The heatmap in B1 B2 visually underscores the superior efficacy of BUM30 + MEB1, with a clear gradient shift towards the darker spectrum, indicative of higher cytotoxicity and morphological alterations. In contrast, BUM30 alone exhibited only moderate effects, aligning with its primary mechanism targeting hyperexcitability rather than direct cytotoxicity.
The box plot in C1 C2 consolidates the distribution of final scores, quantitatively reinforcing the pronounced efficacy of the combination treatment across all tumoroids (****p < 0.0001). The highest median final scores were consistently observed in the BUM30 + MEB1 condition emphasizing the enhanced cytotoxicity achieved through combination therapy.
Finally, the bar plots in D1 D2 illustrate the median final scores. BUM30 + MEB1 consistently outperformed all other conditions, reaching near-maximal scores indicative of complete cell disruption and apoptosis.
Overall, these findings highlight the robust synergistic effect of BUM30 + MEB1 in inducing both cytotoxicity and pronounced morphological damage. The integration of both viability and morphological assessments into a single final score provides a comprehensive framework for evaluating therapeutic efficacy under variable tumor conditions.
4. Discussion
Results validate the advantages of combining MEB and BUM to treat BTs. BUM and MEB act on different mechanisms - ionic distribution and cell volume on one hand and cytoskeletal proteins on the other. Yet, un-expectedly, they share some common actions on NKCC1 notably indirect complex actions of MEB on NKCC1 of human cell lines. In rodent neurons, the combo augments BUM's efficacy by significantly reducing the dosage of BUM needed to block hyperexcitability. In human BTs, the reversal potential of GABA is depolarizing attesting to high (Cl
-)
i levels and NKCC1 hyperactivity. BUM blocks ongoing epileptiform activity in humans BTs, in keeping with extensive literature showing that BUM restores GABAergic inhibition in brain development & in many pathological conditions where it is depolarizing/excitatory
| [11] | Ben-Ari, Y. NKCC1 Chloride Importer Antagonists Attenuate Many Neurological and Psychiatric Disorders. Trends Neurosci. 40, 536-554 (2017). |
| [12] | Savardi, A., Borgogno, M., De Vivo, M. & Cancedda, L. Pharmacological tools to target NKCC1 in brain disorders. Trends Pharmacol. Sci. 42, 1009-1034 (2021). |
[11, 12]
. MEB produces massive apoptosis with up to almost complete cell loss in freshly removed tumors from patients. This effect is time dependent, being more conspicuous 48 hrs. than after 24hrs and even more so after 5 days (
Figures 5, 6). In Neuro
Gfp GBM
mcherry/fLuc cultures, the combo is more efficient in producing cell death than independent applications further attesting their complementary actions. Both drugs are generic being used for decades, thereby facilitating their use in clinical trials. In addition, the duality of their actions reduces side effects that are in literally absent at the sub micromolar dosages used. In sum, this preclinical study justifies the use of the combo in clinical trials and its ads versus MEB or BUM alone.
The following points deserve emphasis concerning GABA, NKCC1 and BUM. GABA
A receptors are present in astrocytoma, oligo-dendroglial tumor
| [29] | Smits, A. et al. GABA-a channel subunit expression in human glioma correlates with tumor histology and clinical outcome. PLoS One 7, 1-10 (2012). |
[29]
and GBM
. GABA receptors participate in the proliferation and migration of these cancer cells. H3K27M+ DMG cells broadly expressed GABA
A receptor (GABA
AR) subunit genes, including α-subunits, β-subunits and γ-subunits, as well as GPHN (encoding gephyrin), ARHGEF9 and NLGN2, which are associated with GABAergic postsynaptic compartments. These genes were expressed to a much greater extent in H3K27M+ DMG and IDH-mutant high-grade gliomas than in IDH WT high-grade gliomas
| [30] | Ravi, V. M. et al. Spatially resolved multi-omics deciphers bidirectional tumor-host interdependence in glioblastoma. Cancer Cell 40, 639-655. e13 (2022). |
| [53] | Berger, F., Lahrech, H. & Ben-ari, Y. Treating Aggressive Brain Tumors with the Combo Bumetanide / Mebendazole : A New Cytotoxic, Anti-Invasive Network Strategy. 10, (2025). |
[30, 53]
. Also, tumors are invaded by axons and a complex network is formed
32 that generate detrimental hyperactivity including excitatory GABA actions due to high (Cl
-)
i levels
Here, we used single GABA channel recordings - the most reliable technique to determine endogenous (Cl
-)
i levels and GABA receptor actions. Interestingly, neurons endowed with immature properties -like high (Cl
-)
i levels and depolarizing actions of GABA - play an important role in many other disorders
| [11] | Ben-Ari, Y. NKCC1 Chloride Importer Antagonists Attenuate Many Neurological and Psychiatric Disorders. Trends Neurosci. 40, 536-554 (2017). |
| [12] | Savardi, A., Borgogno, M., De Vivo, M. & Cancedda, L. Pharmacological tools to target NKCC1 in brain disorders. Trends Pharmacol. Sci. 42, 1009-1034 (2021). |
| [53] | Berger, F., Lahrech, H. & Ben-ari, Y. Treating Aggressive Brain Tumors with the Combo Bumetanide / Mebendazole : A New Cytotoxic, Anti-Invasive Network Strategy. 10, (2025). |
[11, 12, 53]
and there are many similarities between immature neurons & cancer cells
| [21] | Wang, X., Liang, J. & Sun, H. The Network of Tumor Microtubes: An Improperly Reactivated Neural Cell Network With Stemness Feature for Resistance and Recurrence in Gliomas. Front. Oncol. 12, 1-11 (2022). |
[21]
.
NKCC1 antagonists and notably BUM might be useful to treat a wide range of cancers. The underlying concept is that reduction of cell volume is a hallmark of apoptosis and this is largely dependent on NKCC1 activity
| [7] | Algharabil, J. et al. Inhibition of Na-K + -2Cl - cotransporter isoform 1 accelerates temozolomidemediated apoptosis in glioblastoma cancer cells. Cell. Physiol. Biochem. (2012) https://doi.org/10.1159/000339047 |
[7]
Loss of cell volume and lowering of ionic strength of intracellular K
+ and CL
- occur before detectable signs of apoptosis. Cancer cells like GBM maintain basal [Cl
-]
i levels well above the electrochemical equilibrium of the ion (∼ 68 mM) and NKCC1 acts to replenish [Cl
-]
i levels. Temozolomide -like Bum - triggered cell volume loss and accelerated early apoptosis and activation of caspase3 and 4
| [7] | Algharabil, J. et al. Inhibition of Na-K + -2Cl - cotransporter isoform 1 accelerates temozolomidemediated apoptosis in glioblastoma cancer cells. Cell. Physiol. Biochem. (2012) https://doi.org/10.1159/000339047 |
| [34] | Cuddapah, V. A. & Sontheimer, H. Ion channels and tranporters in cancer. 2. Ion channels and the control of cancer cell migration. Am. J. Physiol. - Cell Physiol. 301, 1-16 (2011). |
| [35] | Luo, L. et al. Blockade of cell volume regulatory protein NKCC1 increases TMZ-induced glioma apoptosis and reduces astrogliosis. Mol. Cancer Ther. (2020) https://doi.org/10.1158/1535-7163.mct-19-0910 |
[7, 34, 35]
Interestingly, NKCC1 is enriched in the leading edge of the migrating cellular element
| [6] | Garzon-Muvdi, T. et al. Regulation of brain tumor dispersal by NKCC1 through a novel role in focal adhesion regulation. PLoS Biol. 10, (2012). |
[6]
. It is also phosphorylated with augmented activity in tumors including GBM and this correlates with the histological grade and severity of GBM
Linking cell volume changes, and mTOR signaling has also been suggested to mediate the effects of bumetanide
| [37] | Demian, W. L. et al. The Ion Transporter NKCC1 Links Cell Volume to Cell Mass Regulation by Suppressing mTORC1. Cell Rep. 27, 1886-1896. e6 (2019). |
[37]
. Bum attenuates a wide range of cancers including hepatic
| [38] | Zhou, Y. et al. Discovery of NKCC1 as a potential therapeutic target to inhibit hepatocellular carcinoma cell growth and metastasis. Oncotarget 8, 66328-66342 (2017). |
[38]
lung
| [39] | Andersson, B., Behnam-Motlagh, P., Henriksson, R. & Grankvist, K. Pharmacological modulation of lung cancer cells for potassium ion depletion. Anticancer Res. 25, 2609-2616 (2005). |
[39]
, pancreatic
| [40] | Guerra, B., Fischer, M., Schaefer, S. & Issinger, O. G. The kinase inhibitor D11 induces caspase-mediated cell death in cancer cells resistant to chemotherapeutic treatment. J. Exp. Clin. Cancer Res. 34, 1-14 (2015). |
[40]
, and colon cancers
| [41] | Malamas, A. S., Jin, E., Zhang, Q., Haaga, J. & Lu, Z. R. Anti-angiogenic Effects of Bumetanide Revealed by DCE-MRI with a Biodegradable Macromolecular Contrast Agent in a Colon Cancer Model. Pharm. Res. 32, 3029-3043 (2015). |
[41]
. Patients with high NKCC1 expression had significantly shorter disease-free survival (DFS; P<0.001) and shorter overall survival (OS; P<0.001)
| [42] | Sun, P. L. et al. Expression of Na+-K+-2Cl- cotransporter isoform 1 (NKCC1) predicts poor prognosis in lung adenocarcinoma and EGFR-mutated adenocarcinoma patients. Qjm 109, 237-244 (2016). |
[42]
. [Cl
-]
i levels are elevated in prostate cancer that modulate cell proliferation
| [43] | Hiraoka, K. et al. Chloride ion modulates cell proliferation of human androgen-independent prostatic cancer cell. Cell. Physiol. Biochem. 25, 379-388 (2010). |
[43]
NKCC1 activity is also correlated in tumor related epilepsies with significant actions of Bumetanide
| [44] | Huberfeld, G. & Vecht, C. J. Seizures and gliomas - Towards a single therapeutic approach. Nat. Rev. Neurol. 12, 204-216 (2016). |
[44]
. NKCC1 activity is involved in the epithelial to mesenchymal transition and as such a prognostic biomarker of GBM severity
| [9] | Sun, H., Long, S., Wu, B., Liu, J. & Li, G. NKCC1 involvement in the epithelial-to-mesenchymal transition is a prognostic biomarker in gliomas. PeerJ 8, 1-14 (2020). |
[9]
. In sum, the strong interactions between the tumors and their environment leads to a high NKCC1 activity, high (Cl
-)
i levels and seizures. The reinforcement of the anti-epileptic actions of Bumetanide by incubation with anti-helminthic agents deserves emphasis, as patients with brain tumors have recurrent seizures necessitating anti-epileptic treatment.
The anthelmintic antiparasitic agent Mebendazole acts by depolymerizing cytoskeletal proteins
| [15] | Bai, R. Y., Staedtke, V., Aprhys, C. M., Gallia, G. L. & Riggins, G. J. Antiparasitic mebendazole shows survival benefit in 2 preclinical models of glioblastoma multiforme. Neuro. Oncol. 13, 974-982 (2011). |
[15]
MEB meets several requirements desirable for a repurposed drug to treat cancers notably GBM
| [45] | Guerini, A. E. et al. Mebendazole as a Candidate for Drug Repurposing in Oncology : An Extensive Review of Current Literature. Cancers (Basel). 1-22 (2019). |
[45]
. Experimental observations suggest an increase of apoptosis
| [46] | Ren, L. et al. Benzimidazoles induce concurrent apoptosis and pyroptosis of human glioblastoma cells via arresting cell cycle. Acta Pharmacol. Sin. 1-15 (2021) https://doi.org/10.1038/s41401-021-00752-y |
| [47] | Kipper, F. C. et al. Vinblastine and antihelmintic mebendazole potentiate temozolomide in resistant gliomas. Invest. New Drugs 36, 323-331 (2018). |
[46, 47]
. Various clinical trials have met some success with a combination of MEB and Temozolomide. Thus, Kaplan -Meier analysis showing a 21 months median overall survival with 41% of patients alive at 2 years
| [18] | Gallia, G. L. et al. Neuro-Oncology Advances diagnosed high-grade gliomas : results of a phase 1. neuro-oncology Adv. 3, 1-8 (2021). |
| [15] | Bai, R. Y., Staedtke, V., Aprhys, C. M., Gallia, G. L. & Riggins, G. J. Antiparasitic mebendazole shows survival benefit in 2 preclinical models of glioblastoma multiforme. Neuro. Oncol. 13, 974-982 (2011). |
[18, 15]
Median progression-free survival from date of diagnosis was of 13, 1 months (also see
| [48] | Meco, D., Attinà, G., Mastrangelo, S., Navarra, P. & Ruggiero, A. Emerging Perspectives on the Antiparasitic Mebendazole as a Repurposed Drug for the Treatment of Brain Cancers. Int. J. Mol. Sci. 24, (2023). |
[48]
and 2 trials with MEB are ongoing (trial.gouv.com). MEB plus radiation provides a better survival benefit beyond either alone in triple negative breast cancer and malignant meningioma
| [49] | zhang L dratver MB yazal T et al. mebendazole potentiates radiation therapy in triple negative breast cancer. int j oncol biol phys 01 103, 195-207 (2019). |
[49]
. In rodent model, malignant of malignant meningioma; MEB enhanced efficacy of radiation
| [50] | Skibinski, C. G., Williamson, T. & Riggins, G. J. Mebendazole and radiation in combination increase survival through anticancer mechanisms in an intracranial rodent model of malignant meningioma. J. Neurooncol. 140, 529-538 (2018). |
[50]
. In a pilot trial, MEB monotherapy achieved long term disease control of metastatic adrenocortical carcinoma
. MEB also disrupt tumorigenesis in pancreatic cancer
17 and MEB or an analogue Flubendazole induces mitotic catastrophe cell in melanoma
| [52] | Rudolf, K. & Rudolf, E. Toxicology in Vitro An analysis of mitotic catastrophe induced cell responses in melanoma cells exposed to fl ubendazole. Toxicol. Vitr. 68, 104930 (2020). |
[52]
. The synergistic actions of both agents on hippocampal neurons suggest that alterations of cytoskeletal proteins alter intrinsic properties AND reinforcing GABAergic effects with concentrations as low as 30nM of bumetanide that have no effects on naïve neurons attenuates seizures and blocks NKCC1 and depolarizing /excitatory actions of GABA. Blocking evoked seizures with such low concentrations of Bumetanide is to the best of our knowledge unprecedented. It is likely that in clinical trials, this will reduce the side effects of both 2 agents, notably the diuretic actions of Bumetanide observed in clinical trials on autism
| [13] | Lemonnier, E. et al. Effects of bumetanide on neurobehavioral function in children and adolescents with autism spectrum disorders. Transl. Psychiatry 7, 1-9 (2017). |
[13]
.
Collectively, these observations suggest that the combo BUM and MEB might be useful to treat meningioma, GBM and brain metastasis. Although acting on different sites and mechanisms, they have a complementary action. In keeping with this, we have performed a pilot compassionate trial on a patient with a large un-operable large brain stem metastatic tumor inducing a radiological response, major clinical response, cortico-steroids reduction and significant increased life survival
| [53] | Berger, F., Lahrech, H. & Ben-ari, Y. Treating Aggressive Brain Tumors with the Combo Bumetanide / Mebendazole : A New Cytotoxic, Anti-Invasive Network Strategy. 10, (2025). |
[53]
. Clearly, more trials are warranted.
This study has several limitations. Firstly, the small number of tumors accessible to investigations strongly limits the conclusions that can be derived from the study. Although due to obvious issues, this problem must be considered. Future studies will have to rely on larger samples to validate our conclusions. Considering the difficulty of obtaining cortical tumors in conditions that are appropriate with electrophysiological and tumoroid preparations, the study is limited in the type & numbers of brain samples. We clearly need more diversified material to ensure representativity and relevance to some or all types of BTs. In addition, conclusions derived from this very heterogenous collection of tumoroids must be validated by further experiments and possibly circumscribed to some types of tumors when sufficient collections of data will be available. The use of different preparations, although important to avoid their intrinsic limitations, pose the problem of comparing their effects. Another limitation stems from the relatively short life span of in vitro tumoroids, reflected by the bigger efficacy of the combo after 48 hours than 24hrs. This limitation however will be alleviated in clinical trials. Nevertheless, the material accumulated in various human and rodent preparations speak in favor of a complementary action of 2 widely different /complementary agents. Considering the heterogeneity of BTs and the important dynamic plasticity of synaptic interactions with the environment and the resulting hyperactivity, the combination of drugs might in the future be unavoidable to deal with these very different targets and aims. It is for instance likely that patient immunotherapy might have to include other agents to act on the environment. In that respect, present paper paves the way to possible interventions aiming the tumor AND its environment.
Our results add new data connected to the new identified pro-tumoral impact of neuronal activity on brain tumor. For the first time, we demonstrate the involvement of NKCC1 in this major glioma driver and the connected impact of the Bum/MEB combo. Moreover, dual neuron-glioblastoma experiments, suggest that the neuronal driver also inhibits tumor cell death and that its inhibition using bum restores response to cytotoxic therapy. If we add the already demonstrated anti-invasive impact of Bum, this provides a new multi-targets therapy targeting both neuronal micro-environment and the tumor itself.
To date no anti-invasive therapy is available, and individualization of the protumor neuronal activity was not translated at the bedside in a specific drug therapy. The referent Temozolomide therapy for glioblastoma induces neurotoxicity probably increasing the neuronal protumor activity and enhancing tumor invasion/migration. Bum/Mem combo would inhibit both, providing for the first time a drug combination targeting two major drivers of brain tumor aggressiveness. Finally, the concept that emerges from the present and many recent studies is that BT cells and their immediate environment have immature features that must be considered in a cognitive and therapeutic perspective.
Figure 1. BUM and MEB fully and partly block NKCC1 respectively.
Figure 1. Effect of MBD on NKCC1 function. A. Effect of ouabain (200 µM), bumetanide (20 µM), and both on K
+ influx in HEK293 cells. B. Effect of MEB (0.1 - 30 µM) on ouabain-resistant K
+ influx. C. Effect of MEB (0.1 - 31.6 µM) applied during pre-incubation (15 min) and flux (15 min) on ouabain-resistant K
+ flux. D. Effect of MEB (10 µM during pre-incubation and flux) at different low bumetanide concentrations (0, 60 and 200 nM). Bars represent means ± SEM (n = 3).
Figure 2. Synergistic actions of benzimidazoles and BUM on inhibition of GABAergic Giant Depolarizing Potentials (GDP) in neonatal hippocampal slices.
A, B GDP recorded in Voltage- Clamp (Vc) from CA3 hippocampal neurons. Note that MEB (20 µM) did not alter their frequency. However, both in naïve slices (A) and after incubation with MEB (20uM) for 1 hour (B), bumetanide fully blocked the GDP (A-C). C, Quantitative measures. D-F, Mebendazole enhances potency of bumetanide inhibition. Representative Vc recordings of GDPs in control and in the presence of 30 nM of bumetanide in a control slice (D) and in a mebendazole treated slice (E). Low concentration of bumetanide (30 nM) inhibits GDPs activity in hippocampal slices treated with mebendazole (E, F) but not in control (D, F). F, Quantitative measures. The error bars represent SEM.
Figure 3. Bumetanide in combination with mebendazole produces anti-epileptic activity.
A, B,- In peritumoral human GBM cortex, pyramidal neurons (A) exhibit ongoing epileptiform activity. Action potential firing pattern of neuron is shown in insert. B, Representative traces of current-clamp patch-clamp recordings of epileptiform activity from pyramidal neuron. C, Bumetanide (2 µM) blocks the paroxysmal activity.
D: In rodent hippocampal slices, high frequency tetanus (20 stimuli, 100 Hz) produces long lasting episodes of Interictal activity. This epileptiform was fully blocked by bumetanide (D, G, H) or co-application of MEB (20 µM) and BUM (2 µM) (E, G, H) but it was not altered by MEB (20 µM) alone (E, G, H). Representative superimposed traces of tetanus-induced seizures in control condition, bumetanide (D), in mebendazole and combination of mebendazole and bumetanide (E) and corresponding averaged frequency histograms (F, G). H: Quantified data on 5 cells. Note that applications of mebendazole (20 µM) had little effects on spikes counts, whereas Bum or co-applications of MEB and BUM fully blocked seizure-like activity. The error bars represent SEM.
Figure 4. Cell death produced by the combo in NeuroGfpGBMmcherry/fLuc co-cultures.
(A) Left: Brightfield and fluorescent representative images of NeuroGfpGBMmcherry/fLuc co-cultures 72 h after treatment. 10X magnification with Z-stack. Scale bar 100 m. Right: Tumor area calculated from red fluorescent area of NeuroGfpGBMmcherry/fLuc co-cultures. (B) Percentage of tumor dead cells was determined by DRAQ7 staining on mCherry positive cells (glioblastoma) after NeuroGfpGBMmcherry/fLuc culture dissociation. (C) Caspase-3 activity in NeuroGfpGBMmcherry/fLuc cultures. Caspase-3 activity was normalized on fLuciferase (which is expressed only in glioblastoma cells) after 72 hours of treatment.
Figure 5. Viability Assay Using Live/Dead Kit on Meningioma Tumoroids.
Effect of bumetanide (B 30 µM), mebendazole (M; 0.5 µM, 1 µM), and their combinations on meningioma cell viability in tumoroids-on-chip derived from five patients. The ratio of dead cells to total cells was quantified using a Live/Dead fluorescence assay at 24 h and/or 48 h. Data represent mean ± SD from triplicate chips per condition. Statistical significance was assessed using one-way ANOVA with Tukey’s multiple comparisons test. see text.
Figure 6. Integrated evaluation of Bumetanide and Mebendazole effects on brain tumoroids: A 5-Days treatment assessment.
A1 A2 Quantitative assessment of 16 brain tumoroid models treated with Bumetanide (B, 30 µM), Mebendazole (M, 1 µM), and their combination (B 30 µM + M 1 µM) for 5 days. Viability was evaluated using a dye exclusion assay with propidium iodide and calcein, scoring from 0 to 4: 0 = no cell death (0%), 1 = low cell death (<25%), 2 = moderate (25-50%), 3 = high (50-75%), 4 = near-complete cell death (>75%). Morphological alterations were quantified by phase-contrast microscopy and ImageJ analysis, based on nuclear condensation, cytoskeletal disorganization, and cell shrinkage, scored from 0 to 3: 0 = no change, 1 = mild alterations, 2 = moderate changes (e.g., rounded cells, partial cytoskeletal collapse), 3 = severe morphological damage (condensed, fragmented, apoptotic cells). The Final Score represents the sum of viability and morphological scores.
B1 B2 Heatmap representation of final scores across all tumoroids and treatments. Conditions are plotted on the x-axis, with individual tumoroids on the y-axis. A grayscale color scale was applied (white = low final score, black = high final score). This visualization reveals the superior efficacy of the B + M combination.
Brief Key Points
1) Combined actions of Meb and BUM block brain tumors hyperactivity, while enhancing apoptosis.
2) Large clinical trials are warranted to evaluate the potential benefit of this combined treatment.
Brief Points
1) Co-administration of a NKCC1 inhibitor and a microtubule destructor enhances their anticancer activities.
2) Use of this combo of 2 repositioned generic drugs is recommended to treat brain tumors.
Importance of the Study
Recent studies indicate that brain tumors establish connections with its environment generating hyperactivity that enhance metastasis & aggravate the prognosis suggesting that treatments must exert a dual action on the tumor and its environment. We report that 2 generic repositioned anticancer drugs (Bumetanide & Mebendazole) have complementary and synergistic actions on brain tumors. In freshly recorded tumors, single GABA channel patch recordings suggest high (Cl-)i levels attesting to an overactivity of the co-transporter NKCC1. The NKCC1 inhibitor Bumetanide reduces (Cl-)i levels, restores GABAergic inhibition & blocks ongoing interictal activity. In rodent hippocampal slices, Mebendazole augments four folds the efficacy of co-applied Bumetanide. In brain tumoroids extracted from patients & in mixed brain/GBM cultures, micromolar concentrations of Mebendazole coupled with Bumetanide produced significant apoptosis and fully blocked neuronal hyperactivity. These observations call for large clinical tests using the combo that augment anti-cancer actions while reducing side effects.
Abbreviations
GDP | Giant Depolarizing Potentials |
MEB | Mebnedazole |
ALB | Albendazole |
BUM | Bumetanide |
NKCC1 | Sodium, Potassium, Chloride Co-transporter |
(Cl-)i | Intracellular Chloride Levels |
GBM | Glioblastoma |
TMZ | Temozolomide |
GPSCs | GABA Post Synaptic Currents |
DF GABA | Driving Force of GABAergic Currents |
GABA aR | GABA Receptors |
GPHN | Gephyrin |
GFP | Green Fluorescent Protein |
Author Contributions
William Bourgeois: Investigation, Methodology
Natalia Lozovaya: Investigation, Methodology, Supervision, Writing – review & editing
Marianna Silvano: Investigation, Methodology, Writing – review & editing
Anice Moumen: Investigation, Methodology, Writing – review & editing
Eric Delpire: Data curation, Investigation, Methodology, Writing – review & editing
Arnaud Lazard: Investigation, Methodology, Writing – review & editing
Dominique Hoffman: Investigation, Methodology, Writing – review & editing
Nikolai Zukovsky: Investigation, Methodology, Project administration, Supervision, Validation, Writing – review & editing
Francois Berger: Funding acquisition, Project administration, Resources, Supervision, Writing – review & editing
Yehezkel Ben-Ari: Conceptualization, Funding acquisition, Investigation, Project administration, Supervision, Validation, Writing – original draft, Writing – review & editing
Funding
Funding of this work was provided by
a) European Grant to Ba-Oncomedical & Neurix (project number 3411, acronym Chloroblast)
b) ongoing INSERM funds provided to The Brain tech lab (F. Berger)
c) Ongoing financial support from Ba-Oncomedical startup (Y Ben-Ari)
Data Availability Statement
The data will be made available upon reasonable request except for personal data of the patients treated.
Conflicts of Interest
The authors declare no conflicts of interest.
References
| [1] |
Yabo, Y. A., Niclou, S. P. & Golebiewska, A. Cancer cell heterogeneity and plasticity: A paradigm shift in glioblastoma. Neuro. Oncol. 24, 669-682 (2022).
|
| [2] |
Stupp, R. et al. Maintenance therapy with tumor-Treating fields plus temozolomide vs temozolomide alone for glioblastoma a randomized clinical trial. JAMA - J. Am. Med. Assoc. 314, 2535-2543 (2015).
|
| [3] |
White, J., White, M. P. J., Wickremesekera, A., Peng, L. & Gray, C. The tumour microenvironment, treatment resistance and recurrence in glioblastoma. J. Transl. Med. 22, 1-14 (2024).
|
| [4] |
A Roncevic, N Koruga, A S Koriga, R. R. Why do Glioblastoma treatments fail? Futur. Pharmacol. 5, 1-16 (2025).
|
| [5] |
Stupp R et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma A Randomized Clinical Trial. 318, 2306-2316 (2017).
|
| [6] |
Garzon-Muvdi, T. et al. Regulation of brain tumor dispersal by NKCC1 through a novel role in focal adhesion regulation. PLoS Biol. 10, (2012).
|
| [7] |
Algharabil, J. et al. Inhibition of Na-K + -2Cl - cotransporter isoform 1 accelerates temozolomidemediated apoptosis in glioblastoma cancer cells. Cell. Physiol. Biochem. (2012)
https://doi.org/10.1159/000339047
|
| [8] |
Kourghi, M., Pei, J. V., De Ieso, M. L., Flynn, G. & Yool, A. J. Bumetanide derivatives AqB007 and AqB011 selectively block the aquaporin-1 ion channel conductance and slow cancer cell migration. Mol. Pharmacol. 89, 133-140 (2016).
|
| [9] |
Sun, H., Long, S., Wu, B., Liu, J. & Li, G. NKCC1 involvement in the epithelial-to-mesenchymal transition is a prognostic biomarker in gliomas. PeerJ 8, 1-14 (2020).
|
| [10] |
Ma, H. et al. NKCC1 promotes EMT-like process in GBM via RhoA and Rac1 signaling pathways. J. Cell. Physiol. (2019)
https://doi.org/10.1002/jcp.27033
|
| [11] |
Ben-Ari, Y. NKCC1 Chloride Importer Antagonists Attenuate Many Neurological and Psychiatric Disorders. Trends Neurosci. 40, 536-554 (2017).
|
| [12] |
Savardi, A., Borgogno, M., De Vivo, M. & Cancedda, L. Pharmacological tools to target NKCC1 in brain disorders. Trends Pharmacol. Sci. 42, 1009-1034 (2021).
|
| [13] |
Lemonnier, E. et al. Effects of bumetanide on neurobehavioral function in children and adolescents with autism spectrum disorders. Transl. Psychiatry 7, 1-9 (2017).
|
| [14] |
Florio, R. et al. Screening of benzimidazole-based anthelmintics and their enantiomers as repurposed drug candidates in cancer therapy. Pharmaceuticals 14, 1-16 (2021).
|
| [15] |
Bai, R. Y., Staedtke, V., Aprhys, C. M., Gallia, G. L. & Riggins, G. J. Antiparasitic mebendazole shows survival benefit in 2 preclinical models of glioblastoma multiforme. Neuro. Oncol. 13, 974-982 (2011).
|
| [16] |
Simbulan-rosenthal, C. M. et al. The repurposed anthelmintic mebendazole in combination with trametinib suppresses refractory NRAS Q61K melanoma. 8, 12576-12595 (2017).
|
| [17] |
Williamson, T. et al. Mebendazole disrupts stromal desmoplasia and tumorigenesis in two models of pancreatic cancer. Oncotarget 12, 1326-1338 (2021).
|
| [18] |
Gallia, G. L. et al. Neuro-Oncology Advances diagnosed high-grade gliomas : results of a phase 1. neuro-oncology Adv. 3, 1-8 (2021).
|
| [19] |
Losada-Pérez, M., García-Moreno, M. H., García-Ricote, I. & Casas-Tintó, S. Synaptic components are required for glioblastoma progression in Drosophila. PLoS Genet. 18, 1-27 (2022).
|
| [20] |
Venkatesh, H. S. et al. Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature (2017)
https://doi.org/10.1038/nature24014
|
| [21] |
Wang, X., Liang, J. & Sun, H. The Network of Tumor Microtubes: An Improperly Reactivated Neural Cell Network With Stemness Feature for Resistance and Recurrence in Gliomas. Front. Oncol. 12, 1-11 (2022).
|
| [22] |
Barron, T. et al. GABAergic neuron-to-glioma synapses in diffuse midline gliomas. Nature (2025)
https://doi.org/10.1038/s41586-024-08579-3
|
| [23] |
Koumangoye, R., Penny, P. & Delpire, E. Loss of NKCC1 function increases epithelial tight junction permeability by upregulating claudin-2 expression. Am. J. Physiol. - Cell Physiol. 323, C1251-C1263 (2022).
|
| [24] |
Lozovaya, N. et al. Early alterations in a mouse model of Rett syndrome: the GABA developmental shift is abolished at birth. Sci. Rep. 9, 1-16 (2019).
|
| [25] |
Ben-Ari, Y., Gaiarsa, J.-L., Tyzio, R. & Khazipov, R. GABA: A pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol. Rev. 87, 1215-1268 (2007).
|
| [26] |
Martinez Bedoya, D. et al. PTPRZ1-Targeting RNA CAR T Cells Exert Antigen-Specific and Bystander Antitumor Activity in Glioblastoma. Cancer Immunol. Res. 12, 1718-1735 (2024).
|
| [27] |
Cosset, É. et al. Human three-dimensional engineered neural tissue reveals cellular and molecular events following cytomegalovirus infection. Biomaterials 53, 296-308 (2015).
|
| [28] |
Ben-Ari, Y., Cherubini, E., Corradetti, R. & Gaiarsa, J. L. Giant synaptic potentials in immature rat CA3 hippocampal neurones. J. Physiol. 416, 303-325 (1989).
|
| [29] |
Smits, A. et al. GABA-a channel subunit expression in human glioma correlates with tumor histology and clinical outcome. PLoS One 7, 1-10 (2012).
|
| [30] |
Ravi, V. M. et al. Spatially resolved multi-omics deciphers bidirectional tumor-host interdependence in glioblastoma. Cancer Cell 40, 639-655. e13 (2022).
|
| [31] |
Ren, Y. et al. Spatial transcriptomics reveals niche-specific enrichment and vulnerabilities of radial glial stem-like cells in malignant gliomas. Nat. Commun. 14, (2023).
|
| [32] |
Cuddapah, V. A., Robel, S., Watkins, S. & Sontheimer, H. A neurocentric perspective on glioma invasion. Nat. Rev. Neurosci. 15, 455-465 (2014).
|
| [33] |
Ben-Ari, Y. Excitatory actions of GABA during development: The nature of the nurture. Nat. Rev. Neurosci. 3, 728-739 (2002).
|
| [34] |
Cuddapah, V. A. & Sontheimer, H. Ion channels and tranporters in cancer. 2. Ion channels and the control of cancer cell migration. Am. J. Physiol. - Cell Physiol. 301, 1-16 (2011).
|
| [35] |
Luo, L. et al. Blockade of cell volume regulatory protein NKCC1 increases TMZ-induced glioma apoptosis and reduces astrogliosis. Mol. Cancer Ther. (2020)
https://doi.org/10.1158/1535-7163.mct-19-0910
|
| [36] |
Zhu, W. et al. WNK1-OSR1 kinase-mediated phospho-activation of Na+-K+-2Cl- cotransporter facilitates glioma migration. Mol. Cancer (2014)
https://doi.org/10.1186/1476-4598-13-31
|
| [37] |
Demian, W. L. et al. The Ion Transporter NKCC1 Links Cell Volume to Cell Mass Regulation by Suppressing mTORC1. Cell Rep. 27, 1886-1896. e6 (2019).
|
| [38] |
Zhou, Y. et al. Discovery of NKCC1 as a potential therapeutic target to inhibit hepatocellular carcinoma cell growth and metastasis. Oncotarget 8, 66328-66342 (2017).
|
| [39] |
Andersson, B., Behnam-Motlagh, P., Henriksson, R. & Grankvist, K. Pharmacological modulation of lung cancer cells for potassium ion depletion. Anticancer Res. 25, 2609-2616 (2005).
|
| [40] |
Guerra, B., Fischer, M., Schaefer, S. & Issinger, O. G. The kinase inhibitor D11 induces caspase-mediated cell death in cancer cells resistant to chemotherapeutic treatment. J. Exp. Clin. Cancer Res. 34, 1-14 (2015).
|
| [41] |
Malamas, A. S., Jin, E., Zhang, Q., Haaga, J. & Lu, Z. R. Anti-angiogenic Effects of Bumetanide Revealed by DCE-MRI with a Biodegradable Macromolecular Contrast Agent in a Colon Cancer Model. Pharm. Res. 32, 3029-3043 (2015).
|
| [42] |
Sun, P. L. et al. Expression of Na+-K+-2Cl- cotransporter isoform 1 (NKCC1) predicts poor prognosis in lung adenocarcinoma and EGFR-mutated adenocarcinoma patients. Qjm 109, 237-244 (2016).
|
| [43] |
Hiraoka, K. et al. Chloride ion modulates cell proliferation of human androgen-independent prostatic cancer cell. Cell. Physiol. Biochem. 25, 379-388 (2010).
|
| [44] |
Huberfeld, G. & Vecht, C. J. Seizures and gliomas - Towards a single therapeutic approach. Nat. Rev. Neurol. 12, 204-216 (2016).
|
| [45] |
Guerini, A. E. et al. Mebendazole as a Candidate for Drug Repurposing in Oncology : An Extensive Review of Current Literature. Cancers (Basel). 1-22 (2019).
|
| [46] |
Ren, L. et al. Benzimidazoles induce concurrent apoptosis and pyroptosis of human glioblastoma cells via arresting cell cycle. Acta Pharmacol. Sin. 1-15 (2021)
https://doi.org/10.1038/s41401-021-00752-y
|
| [47] |
Kipper, F. C. et al. Vinblastine and antihelmintic mebendazole potentiate temozolomide in resistant gliomas. Invest. New Drugs 36, 323-331 (2018).
|
| [48] |
Meco, D., Attinà, G., Mastrangelo, S., Navarra, P. & Ruggiero, A. Emerging Perspectives on the Antiparasitic Mebendazole as a Repurposed Drug for the Treatment of Brain Cancers. Int. J. Mol. Sci. 24, (2023).
|
| [49] |
zhang L dratver MB yazal T et al. mebendazole potentiates radiation therapy in triple negative breast cancer. int j oncol biol phys 01 103, 195-207 (2019).
|
| [50] |
Skibinski, C. G., Williamson, T. & Riggins, G. J. Mebendazole and radiation in combination increase survival through anticancer mechanisms in an intracranial rodent model of malignant meningioma. J. Neurooncol. 140, 529-538 (2018).
|
| [51] |
Irina Dobrosotskaya, Gary hammer, David Schteingart, K. maturen and francis W. Case Report. Endocr. Pract. 59-62 (2011)
https://doi.org/10.4158/EP10390.CR
|
| [52] |
Rudolf, K. & Rudolf, E. Toxicology in Vitro An analysis of mitotic catastrophe induced cell responses in melanoma cells exposed to fl ubendazole. Toxicol. Vitr. 68, 104930 (2020).
|
| [53] |
Berger, F., Lahrech, H. & Ben-ari, Y. Treating Aggressive Brain Tumors with the Combo Bumetanide / Mebendazole : A New Cytotoxic, Anti-Invasive Network Strategy. 10, (2025).
|
Cite This Article
-
APA Style
Bourgeois, W., Lozovaya, N., Silvano, M., Moumen, A., Delpire, E., et al. (2025). Dual Targeting of Brain Tumors and Their Environment by Bumetanide and Mebendazole. Cancer Research Journal, 13(3), 98-122. https://doi.org/10.11648/j.crj.20251303.12
 Copy
|
Copy
|
 Download
Download
ACS Style
Bourgeois, W.; Lozovaya, N.; Silvano, M.; Moumen, A.; Delpire, E., et al. Dual Targeting of Brain Tumors and Their Environment by Bumetanide and Mebendazole. Cancer Res. J. 2025, 13(3), 98-122. doi: 10.11648/j.crj.20251303.12
Copy
|
Download
AMA Style
Bourgeois W, Lozovaya N, Silvano M, Moumen A, Delpire E, et al. Dual Targeting of Brain Tumors and Their Environment by Bumetanide and Mebendazole. Cancer Res J. 2025;13(3):98-122. doi: 10.11648/j.crj.20251303.12
Copy
|
Download
-
@article{10.11648/j.crj.20251303.12,
author = {William Bourgeois and Natalia Lozovaya and Marianna Silvano and Anice Moumen and Eric Delpire and Emmanuel Gay and Florian Le Lann and Arnaud Lazard and Dominique Hoffman and Nikolay Zhukovsky and François Berger and Yehezkel Ben-Ari},
title = {Dual Targeting of Brain Tumors and Their Environment by Bumetanide and Mebendazole
},
journal = {Cancer Research Journal},
volume = {13},
number = {3},
pages = {98-122},
doi = {10.11648/j.crj.20251303.12},
url = {https://doi.org/10.11648/j.crj.20251303.12},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.crj.20251303.12},
abstract = {Background: The anti-epileptic NKCC1 inhibitor Bumetanide (BUM) and the microtubule acting anthelminthic agent Mebendazole (MEB), have anti-cancer properties. Tumor & its environment generate neuronal hyperactivity that aggravates the clinical outcome suggesting that their combination might block hyperactivity (BUM) and augment cell death (MEB). Methods: We tested the effects of the combo on i) NKCC1 activity in human cell lines, ii) electrical activity recorded from mouse hippocampal neurons & tumors freshly resected from patients, iii) glioblastoma-brain co-cultures, iv) cell death in human tumoroids. Results: BUM efficiently inhibited NKCC1 & unexpectedly, MEB applications also via a likely indirect action. In rodent hippocampal neurons, BUM blocked GABAergic Giant Depolarizing Potentials and seizures and co-applications of MEB produced a fourfold increase of BUM's efficacy. In freshly removed brain tumors, E GABA reversal recorded with single GABA channels was highly depolarized (close to -25 mV) in keeping with NKCC1 over activity. BUM fully blocked ongoing epileptic activity. In GBM-Brain cultures, the combo produced stronger effects then independent applications of MEB or BUM. In tumoroids, the combo also efficiently produced strong cell death & morphological changes in some tumors. Conclusion: The combination of BUM & MEB acts complementarily on brain tumors, the former blocking seizures, and the latter producing cell death. Their combination increases their hyperactivity inhibitory actions and cell death. The combo might therefore be used to treat brain tumors combining 2 different mechanisms and targets.},
year = {2025}
}
Copy
|
Download
-
TY - JOUR
T1 - Dual Targeting of Brain Tumors and Their Environment by Bumetanide and Mebendazole
AU - William Bourgeois
AU - Natalia Lozovaya
AU - Marianna Silvano
AU - Anice Moumen
AU - Eric Delpire
AU - Emmanuel Gay
AU - Florian Le Lann
AU - Arnaud Lazard
AU - Dominique Hoffman
AU - Nikolay Zhukovsky
AU - François Berger
AU - Yehezkel Ben-Ari
Y1 - 2025/07/07
PY - 2025
N1 - https://doi.org/10.11648/j.crj.20251303.12
DO - 10.11648/j.crj.20251303.12
T2 - Cancer Research Journal
JF - Cancer Research Journal
JO - Cancer Research Journal
SP - 98
EP - 122
PB - Science Publishing Group
SN - 2330-8214
UR - https://doi.org/10.11648/j.crj.20251303.12
AB - Background: The anti-epileptic NKCC1 inhibitor Bumetanide (BUM) and the microtubule acting anthelminthic agent Mebendazole (MEB), have anti-cancer properties. Tumor & its environment generate neuronal hyperactivity that aggravates the clinical outcome suggesting that their combination might block hyperactivity (BUM) and augment cell death (MEB). Methods: We tested the effects of the combo on i) NKCC1 activity in human cell lines, ii) electrical activity recorded from mouse hippocampal neurons & tumors freshly resected from patients, iii) glioblastoma-brain co-cultures, iv) cell death in human tumoroids. Results: BUM efficiently inhibited NKCC1 & unexpectedly, MEB applications also via a likely indirect action. In rodent hippocampal neurons, BUM blocked GABAergic Giant Depolarizing Potentials and seizures and co-applications of MEB produced a fourfold increase of BUM's efficacy. In freshly removed brain tumors, E GABA reversal recorded with single GABA channels was highly depolarized (close to -25 mV) in keeping with NKCC1 over activity. BUM fully blocked ongoing epileptic activity. In GBM-Brain cultures, the combo produced stronger effects then independent applications of MEB or BUM. In tumoroids, the combo also efficiently produced strong cell death & morphological changes in some tumors. Conclusion: The combination of BUM & MEB acts complementarily on brain tumors, the former blocking seizures, and the latter producing cell death. Their combination increases their hyperactivity inhibitory actions and cell death. The combo might therefore be used to treat brain tumors combining 2 different mechanisms and targets.
VL - 13
IS - 3
ER -
Copy
|
Download