Alagille syndrome (ALGS) is a rare autosomal dominant multisystem disorder characterized by marked phenotypic variability, and the diagnosis is sometimes very challenging. We describe a 26-year-old man who presented with progressive jaundice, pruritus, and weight loss over three months, with a history of similar episodes in early childhood. He had previously been diagnosed with atrial septal defect with severe pulmonary stenosis, myopia, and hypothyroidism. After a thorough Clinical examination which revealed icterus, pedal edema, stigmata of chronic liver disease, and characteristic facial dysmorphism with café-au-lait spots, a possibility of Alagille syndrome was considered. Laboratory evaluation showed cholestatic liver dysfunction and pancytopenia. Whole-exome sequencing identified a likely pathogenic JAG1 variant, and liver biopsy confirmed bile duct paucity. This case highlights the diagnostic challenges of ALGS and underscores the importance of meticulous clinical evaluation and comprehensive longitudinal history-taking, as multiple healthcare encounters preceded the correct diagnosis as far as this patient is concerned.
This is an Open Access article, distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is properly cited.
Alagille syndrome (ALGS) is a genetically heterogeneous, autosomal dominant disorder with multisystem involvement and an estimated incidence of 1 in 30,000 live births
[1]
Spinner NB, Loomes KM, Krantz ID, Gilbert MA. Alagille Syndrome. In: Adam MP, Bick S, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2026. Updated January 4, 2024. Available from:
. It is also known as arteriohepatic dysplasia. ALGS results from mutations in the JAG1 gene (98%) or NOTCH2 gene (2%), both of which play roles in the Notch signaling pathway
[2]
Krantz ID, Colliton RP, Genin A, Rand EB, Li L, Piccoli DA, Spinner NB. Spectrum and frequency of jagged1 (JAG1) mutations in Alagille syndrome patients and their families. Am J Hum Genet. 1998; 62(6): 1361–1369.
. ALGS was initially described as a hepatic disorder, but many patients with genetic mutations do not show overt liver disease.
Diagnosis is typically based on the presence of at least three of five major clinical features: butterfly vertebrae, congenital cardiac defects (most commonly involving the pulmonary arteries), posterior embryotoxon, cholestasis with bile duct paucity, and characteristic facial features, with vascular and renal anomalies also reported
[3]
Saleh M, Kamath BM, Chitayat D. Alagille syndrome: clinical perspectives. Appl Clin Genet. 2016; 9: 75–82.
. The hallmark hepatic feature is bile duct paucity, leading to cholestasis
[4]
Danks DM, Campbell PE, Jack I, Rogers JG, Smith AL. Studies of the aetiology of neonatal hepatitis and biliary atresia. Arch Dis Child. 1977; 52(5): 360–367.
Traditional diagnostic criteria for ALGS include histological evidence of bile duct paucity and three of the following features:
2.1. Cholestasis
Cholestasis often presents in infancy, with jaundice, pruritus, xanthomas, and failure to thrive due to fat malabsorption. Liver biopsy may show early ductal proliferation but typically reveals bile duct paucity, with an interlobular bile duct-to-portal tract ratio <0.4
[5]
Feldman M, Friedman lawrence s, Brandt lawrence j. Sleisenger and Fordtran’s. 11th ed. Elsevier; 2021. 1-2206 p.
[5]
. Approximately 15% of patients progress to chronic liver disease and may require liver transplantation
[6]
Emerick KM, Rand EB, Goldmuntz E, Krantz ID, Spinner NB, Piccoli DA. Features of Alagille syndrome in 92 patients: Frequency and relation to prognosis. Hepatology. 1999; 29(3): 822–829.
Posterior embryotoxon is present in 78–89% of patients. Other findings include Axenfeld- Rieger anomaly (corectopia, polycoria), optic disc drusen, and retinal pigment changes
[7]
Hingorani M, Nischal KK, Davies A, Bentley C, Vivian A, Baker AJ, Mieli‑Vergani G, Bird AC, Aclimandos WA. Ocular abnormalities in Alagille syndrome. Ophthalmology. 1999; 106(2): 330–337.
Broad and Prominent forehead, frontal bossing, prominent or large ears, deep-set eyes, hypertelorism, pointed chin and straight or bulbous nose giving a characteristic inverted triangular shaped face. These traits are strongly associated with JAG1 mutations
[8]
Lin HC, Le Hoang P, Hutchinson A, Chao G, Gerfen J, Loomes KM, Krantz I, Kamath BM, Spinner NB. Alagille syndrome in a Vietnamese cohort: mutation analysis and assessment of facial features. Am J Med Genet Part A. 2012; 158A(5): 1005–1013.
Cardiac anomalies are seen in up to 97% of ALGS patients. Pulmonary artery stenosis (branch or peripheral) is most common. Complex defects like Tetralogy of Fallot, aortic coarctation, and septal defects (Atrial Septal Defect (ASD), Ventricular Septal Defect (VSD)) are also reported.
2.5. Skeletal Anomalies
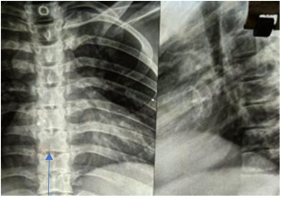
Butterfly vertebrae, due to midline clefting of thoracic vertebral bodies, are seen in 33–93% of cases
[9]
Sanderson E, Newman V, Haigh SF, Baker A, Sidhu PS. Vertebral anomalies in children with Alagille syndrome: an analysis of 50 consecutive patients. Pediatr Radiol. 2002; 32(2): 114–119.
. Other skeletal features may include tapering phalanges and supernumerary flexion creases
[10]
Kamath BM, Loomes KM, Oakey RJ, Krantz ID. Supernumerary digital flexion creases: an additional clinical manifestation of Alagille syndrome. Am J Med Genet. 2002; 112(2): 171–175.
Include renal and vascular anomalies. Vascular abnormalities include intracranial hemorrhage, renovascular anomalies, and middle aortic syndrome.
[12]
Salem JE, Bruguiere E, Iserin L, Guiochon-Mantel A, Plouin PF. Hypertension and aortorenal disease in Alagille syndrome. J Hypertens. 2012; 30(7): 1300–1306.
Renal anomalies such as small echogenic kidneys, cysts, renal artery stenosis, and renal tubular acidosis occur in up to 39% of patients. Patients’ may be having short stature because of immunodeficiency, repeated infections, inherent bone problems (50–90%), poor growth linked to cholestasis, or a serious heart defect.
Diagnosis can be established clinically or genetically. Liver biopsy is not mandatory if genetic confirmation (JAG1/NOTCH2 mutation) is available.
2.7. Genetics of ALGS
98% of cases are caused by mutations in JAG1 and 2% are caused by mutations in NOTCH2. There are no genotype-phenotype associations between the location of the mutation within the gene or the particular types of JAG1 pathogenic variants with the clinical symptoms of ALGS
[13]
Spinner NB, Colliton RP, Crosnier C, Krantz ID, Hadchouel M, Meunier-Rotival M. Jagged1 mutations in Alagille syndrome. Human mutation vol. 17;1 (2001):18–33.
Figure 1. Multiple café au lait spots, Facial dysmorphism with gradual change in his shape of face from 15 years to 26 years.
A 26-year-old man presented with jaundice, dark urine, and pruritus for three months duration. He had experienced similar episodes at 3 months and 18 months of age, with persistent mild pruritus since childhood. He had no history of gastrointestinal bleeding or ascites.
Past medical history included myopia (diagnosed in 2010), congenital heart disease (ASD with severe pulmonary stenosis), balloon pulmonary valvotomy (2012), and ASD device closure (2016). He was diagnosed with hypothyroidism in 2014. There was no family history of similar illness. His siblings were healthy.
On examination, he was pale, icteric, and had bilateral pedal edema. BMI was 17.6 kg/m². Stigmata of chronic cholestasis and chronic liver disease included scratch marks, shiny nails, and spider nevi. He also had multiple café au lait spots over the trunk. (Figure 1). Hepatosplenomegaly was present; cardiac auscultation revealed a systolic murmur. Facial features were consistent with ALGS. There was gradual change in his shape of face from 15 years to 26 years to give the current characteristic facial dysmorphism.
4. Investigations
CBC showed pancytopenia.
LFT: Cholestatic pattern.
Viral and autoimmune markers were negative.
Serum ceruloplasmin and ferritin were normal.
Peripheral smear: Macrocytic RBCs with target cells.
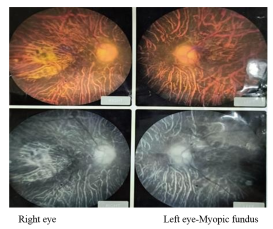
Ophthalmology evaluation: Posterior embryotoxon, high myopia, epiretinal membrane (Figure 2).
Cardiology: Echo showed mild pulmonary stenosis and no residual ASD shunt.
USG: Coarse liver, small echogenic right kidney.
MRCP: Chronic liver disease with portal hypertension.
CECT: Segmental liver atrophy with hypertrophy of segment IV.
Upper GI Endoscopy: Grade III esophageal varices.
Liver biopsy: Bile duct paucity, cholestasis, cirrhosis.
Whole Exome Sequencing: Identified a heterozygous likely pathogenic 5′ splice-site variant c.2458+1G>A in intron 20 of the JAG1 gene (chr20p12.2), aberrant mRNA splicing and functional loss of the Jagged 1 protein consistent with autosomal dominant Alagille syndrome due to JAG1 haploinsufficiency.
This patient had all seven recognized clinical features of Alagille Syndrome: cholestasis, cardiac, ocular, skeletal, renal, vascular abnormalities, and facial dysmorphism. Genetic testing confirmed a pathogenic JAG1 mutation, establishing the diagnosis.
Differentials such as PFIC, autoimmune hepatitis, primary sclerosing cholangitis, and congenital hepatic fibrosis were ruled out based on clinical, histological, and serological findings. Unique findings in this case were café au lait spots and hypothyroidism, not commonly described in ALGS.
Management of ALGS is multidisciplinary. Medical therapy for pruritus includes ursodeoxycholic acid, rifampin, cholestyramine, and naltrexone. In refractory cases, surgical biliary diversion may be considered
[14]
Wang KS, Tiao G, Bass LM, Hertel PM, Mogul D, Kerkar N, Clifton M, Azen C, Bull L, Rosenthal P, Stewart D, Superina R, Arnon R, Bozic M, Brandt ML, Dillon PA, Fecteau A, Iyer K, Kamath B, Karpen SJ, Karrer F, Loomes KM, Mack C, Mattei P, Miethke A, Soltys K, Turmelle YP, West K, Zagory J, Goodhue C, Shneider BL; Childhood Liver Disease Research Network (ChiLDReN). Analysis of surgical interruption of the enterohepatic circulation as a treatment for pediatric cholestasis. Hepatology. 2017; 65(5): 1645–1654.
. Liver transplantation remains the definitive option for progressive liver disease. Cardiac anomalies must also be managed early, as they are a major cause of early mortality. The 5-year survival rate after liver transplantation for ALGS is 80%, and 90% of those afflicted experience some catch-up growth
[15]
Pawlowska J, Socha P, Jankowska I. Factors affecting catch-up growth after liver transplantation in children with cholestatic liver diseases. Ann Transplant. 2010; 15(1): 72–76.
. Genetic counselling is an important component in the management of inherited disorders. The patient was counselled regarding the autosomal dominant inheritance pattern of Alagille syndrome, the possibility of variable clinical expression, the risk of transmission to offspring, and the need for clinical evaluation and genetic testing of family members.
6. Conclusion
Alagille syndrome is a multi-system disorder with highly variable presentation. Genetic testing plays a key role in diagnosis, especially when liver biopsy is not feasible. This case highlights that this rare disorder can remain unrecognized until adulthood due to its heterogenous manifestations. A high index of suspicion with a meticulous history and examination is crucial for timely diagnosis. Increased awareness among clinicians may help prevent diagnostic delays and improve outcome. Multidisciplinary care is essential for optimal outcomes.
Abbreviations
ALGS
Alagille Syndrome
ASD
Atrial Septal Defect
VSD
Ventricular Septal Defect
CBC
Complete Blood Count
LFT
Liver Function Test
RBC
Red Blood Cells
USG
Ultrasonography
MRCP
Magnetic Resonance Cholangiopancreatography
CECT
Contrast Enhanced Computed Tomography
PFIC
Progressive Familial Intrahepatic Cholestasis
Conflicts of Interest
The authors declare no conflicts of interest.
References
[1]
Spinner NB, Loomes KM, Krantz ID, Gilbert MA. Alagille Syndrome. In: Adam MP, Bick S, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2026. Updated January 4, 2024. Available from:
Krantz ID, Colliton RP, Genin A, Rand EB, Li L, Piccoli DA, Spinner NB. Spectrum and frequency of jagged1 (JAG1) mutations in Alagille syndrome patients and their families. Am J Hum Genet. 1998; 62(6): 1361–1369.
Danks DM, Campbell PE, Jack I, Rogers JG, Smith AL. Studies of the aetiology of neonatal hepatitis and biliary atresia. Arch Dis Child. 1977; 52(5): 360–367.
Feldman M, Friedman lawrence s, Brandt lawrence j. Sleisenger and Fordtran’s. 11th ed. Elsevier; 2021. 1-2206 p.
[6]
Emerick KM, Rand EB, Goldmuntz E, Krantz ID, Spinner NB, Piccoli DA. Features of Alagille syndrome in 92 patients: Frequency and relation to prognosis. Hepatology. 1999; 29(3): 822–829.
Lin HC, Le Hoang P, Hutchinson A, Chao G, Gerfen J, Loomes KM, Krantz I, Kamath BM, Spinner NB. Alagille syndrome in a Vietnamese cohort: mutation analysis and assessment of facial features. Am J Med Genet Part A. 2012; 158A(5): 1005–1013.
Sanderson E, Newman V, Haigh SF, Baker A, Sidhu PS. Vertebral anomalies in children with Alagille syndrome: an analysis of 50 consecutive patients. Pediatr Radiol. 2002; 32(2): 114–119.
Kamath BM, Loomes KM, Oakey RJ, Krantz ID. Supernumerary digital flexion creases: an additional clinical manifestation of Alagille syndrome. Am J Med Genet. 2002; 112(2): 171–175.
Salem JE, Bruguiere E, Iserin L, Guiochon-Mantel A, Plouin PF. Hypertension and aortorenal disease in Alagille syndrome. J Hypertens. 2012; 30(7): 1300–1306.
Pawlowska J, Socha P, Jankowska I. Factors affecting catch-up growth after liver transplantation in children with cholestatic liver diseases. Ann Transplant. 2010; 15(1): 72–76.
Perilamthottathil, A., Balagopal, S. K., Kandiyil, S. K. (2026). Unmasking Alagille Syndrome in Adulthood- A Complete Clinical Portrait of a Rare Syndrome. International Journal of Gastroenterology, 10(1), 1-4. https://doi.org/10.11648/j.ijg.20261001.11
Perilamthottathil, A.; Balagopal, S. K.; Kandiyil, S. K. Unmasking Alagille Syndrome in Adulthood- A Complete Clinical Portrait of a Rare Syndrome. Int. J. Gastroenterol.2026, 10(1), 1-4. doi: 10.11648/j.ijg.20261001.11
Perilamthottathil A, Balagopal SK, Kandiyil SK. Unmasking Alagille Syndrome in Adulthood- A Complete Clinical Portrait of a Rare Syndrome. Int J Gastroenterol. 2026;10(1):1-4. doi: 10.11648/j.ijg.20261001.11
@article{10.11648/j.ijg.20261001.11,
author = {Arun Perilamthottathil and Sithara Kodapally Balagopal and Sunil Kumar Kandiyil},
title = {Unmasking Alagille Syndrome in Adulthood- A Complete Clinical Portrait of a Rare Syndrome},
journal = {International Journal of Gastroenterology},
volume = {10},
number = {1},
pages = {1-4},
doi = {10.11648/j.ijg.20261001.11},
url = {https://doi.org/10.11648/j.ijg.20261001.11},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.ijg.20261001.11},
abstract = {Alagille syndrome (ALGS) is a rare autosomal dominant multisystem disorder characterized by marked phenotypic variability, and the diagnosis is sometimes very challenging. We describe a 26-year-old man who presented with progressive jaundice, pruritus, and weight loss over three months, with a history of similar episodes in early childhood. He had previously been diagnosed with atrial septal defect with severe pulmonary stenosis, myopia, and hypothyroidism. After a thorough Clinical examination which revealed icterus, pedal edema, stigmata of chronic liver disease, and characteristic facial dysmorphism with café-au-lait spots, a possibility of Alagille syndrome was considered. Laboratory evaluation showed cholestatic liver dysfunction and pancytopenia. Whole-exome sequencing identified a likely pathogenic JAG1 variant, and liver biopsy confirmed bile duct paucity. This case highlights the diagnostic challenges of ALGS and underscores the importance of meticulous clinical evaluation and comprehensive longitudinal history-taking, as multiple healthcare encounters preceded the correct diagnosis as far as this patient is concerned.},
year = {2026}
}
TY - JOUR
T1 - Unmasking Alagille Syndrome in Adulthood- A Complete Clinical Portrait of a Rare Syndrome
AU - Arun Perilamthottathil
AU - Sithara Kodapally Balagopal
AU - Sunil Kumar Kandiyil
Y1 - 2026/02/14
PY - 2026
N1 - https://doi.org/10.11648/j.ijg.20261001.11
DO - 10.11648/j.ijg.20261001.11
T2 - International Journal of Gastroenterology
JF - International Journal of Gastroenterology
JO - International Journal of Gastroenterology
SP - 1
EP - 4
PB - Science Publishing Group
SN - 2640-169X
UR - https://doi.org/10.11648/j.ijg.20261001.11
AB - Alagille syndrome (ALGS) is a rare autosomal dominant multisystem disorder characterized by marked phenotypic variability, and the diagnosis is sometimes very challenging. We describe a 26-year-old man who presented with progressive jaundice, pruritus, and weight loss over three months, with a history of similar episodes in early childhood. He had previously been diagnosed with atrial septal defect with severe pulmonary stenosis, myopia, and hypothyroidism. After a thorough Clinical examination which revealed icterus, pedal edema, stigmata of chronic liver disease, and characteristic facial dysmorphism with café-au-lait spots, a possibility of Alagille syndrome was considered. Laboratory evaluation showed cholestatic liver dysfunction and pancytopenia. Whole-exome sequencing identified a likely pathogenic JAG1 variant, and liver biopsy confirmed bile duct paucity. This case highlights the diagnostic challenges of ALGS and underscores the importance of meticulous clinical evaluation and comprehensive longitudinal history-taking, as multiple healthcare encounters preceded the correct diagnosis as far as this patient is concerned.
VL - 10
IS - 1
ER -
Perilamthottathil, A., Balagopal, S. K., Kandiyil, S. K. (2026). Unmasking Alagille Syndrome in Adulthood- A Complete Clinical Portrait of a Rare Syndrome. International Journal of Gastroenterology, 10(1), 1-4. https://doi.org/10.11648/j.ijg.20261001.11
Perilamthottathil, A.; Balagopal, S. K.; Kandiyil, S. K. Unmasking Alagille Syndrome in Adulthood- A Complete Clinical Portrait of a Rare Syndrome. Int. J. Gastroenterol.2026, 10(1), 1-4. doi: 10.11648/j.ijg.20261001.11
Perilamthottathil A, Balagopal SK, Kandiyil SK. Unmasking Alagille Syndrome in Adulthood- A Complete Clinical Portrait of a Rare Syndrome. Int J Gastroenterol. 2026;10(1):1-4. doi: 10.11648/j.ijg.20261001.11
@article{10.11648/j.ijg.20261001.11,
author = {Arun Perilamthottathil and Sithara Kodapally Balagopal and Sunil Kumar Kandiyil},
title = {Unmasking Alagille Syndrome in Adulthood- A Complete Clinical Portrait of a Rare Syndrome},
journal = {International Journal of Gastroenterology},
volume = {10},
number = {1},
pages = {1-4},
doi = {10.11648/j.ijg.20261001.11},
url = {https://doi.org/10.11648/j.ijg.20261001.11},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.ijg.20261001.11},
abstract = {Alagille syndrome (ALGS) is a rare autosomal dominant multisystem disorder characterized by marked phenotypic variability, and the diagnosis is sometimes very challenging. We describe a 26-year-old man who presented with progressive jaundice, pruritus, and weight loss over three months, with a history of similar episodes in early childhood. He had previously been diagnosed with atrial septal defect with severe pulmonary stenosis, myopia, and hypothyroidism. After a thorough Clinical examination which revealed icterus, pedal edema, stigmata of chronic liver disease, and characteristic facial dysmorphism with café-au-lait spots, a possibility of Alagille syndrome was considered. Laboratory evaluation showed cholestatic liver dysfunction and pancytopenia. Whole-exome sequencing identified a likely pathogenic JAG1 variant, and liver biopsy confirmed bile duct paucity. This case highlights the diagnostic challenges of ALGS and underscores the importance of meticulous clinical evaluation and comprehensive longitudinal history-taking, as multiple healthcare encounters preceded the correct diagnosis as far as this patient is concerned.},
year = {2026}
}
TY - JOUR
T1 - Unmasking Alagille Syndrome in Adulthood- A Complete Clinical Portrait of a Rare Syndrome
AU - Arun Perilamthottathil
AU - Sithara Kodapally Balagopal
AU - Sunil Kumar Kandiyil
Y1 - 2026/02/14
PY - 2026
N1 - https://doi.org/10.11648/j.ijg.20261001.11
DO - 10.11648/j.ijg.20261001.11
T2 - International Journal of Gastroenterology
JF - International Journal of Gastroenterology
JO - International Journal of Gastroenterology
SP - 1
EP - 4
PB - Science Publishing Group
SN - 2640-169X
UR - https://doi.org/10.11648/j.ijg.20261001.11
AB - Alagille syndrome (ALGS) is a rare autosomal dominant multisystem disorder characterized by marked phenotypic variability, and the diagnosis is sometimes very challenging. We describe a 26-year-old man who presented with progressive jaundice, pruritus, and weight loss over three months, with a history of similar episodes in early childhood. He had previously been diagnosed with atrial septal defect with severe pulmonary stenosis, myopia, and hypothyroidism. After a thorough Clinical examination which revealed icterus, pedal edema, stigmata of chronic liver disease, and characteristic facial dysmorphism with café-au-lait spots, a possibility of Alagille syndrome was considered. Laboratory evaluation showed cholestatic liver dysfunction and pancytopenia. Whole-exome sequencing identified a likely pathogenic JAG1 variant, and liver biopsy confirmed bile duct paucity. This case highlights the diagnostic challenges of ALGS and underscores the importance of meticulous clinical evaluation and comprehensive longitudinal history-taking, as multiple healthcare encounters preceded the correct diagnosis as far as this patient is concerned.
VL - 10
IS - 1
ER -